Neurons in the brain use electrical signals for communicating with each other, and this is the basis for our brain functions such as perception, motor control, learning & memory. But how do neurons acquire excitability during development? What genetic programs control this process? And once neurons acquire the ability to generate electrical signals, they use their activity for circuit formation. But how are intricate neuronal networks in the brain formed by neuronal activity? How genetic program and neuronal activity cooperate to regulate each process of circuit construction? The main focus of our laboratory is to understand molecular, cellular, and network mechanisms underlying activity-dependent circuit formation in the mammalian brain. We use mouse cerebral cortex as a model, and employ state-of-the-art techniques to manipulate gene expression and neuronal activity in developing cortical neurons. Techniques used: mouse genetics, in utero electroporation, patch clump recording, imaging, histology, in vivo activity manipulation and recording.
1. How do neurons in the cerebral cortex acquire excitability during development?
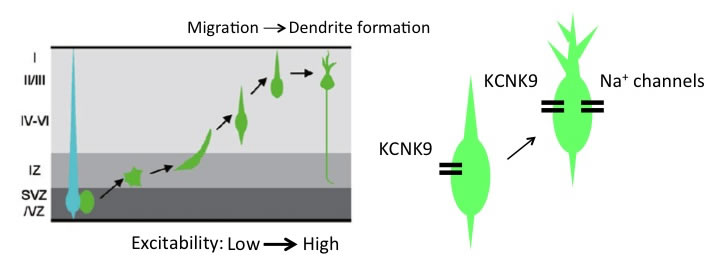
Acquiring the ability to generate action potentials during maturation is a critical developmental step for the functioning of neurons. This occurs around the phase of migration in cortical excitatory neurons in vivo. Migratory neurons show relatively a low level of cellular excitability, and spontaneous activity robustly increases after neurons reach their final positions. Increasing the activity by ectopic expression of a genetic tool NaChBac or inhibition of an endogenously expressed leak K+ channel KCNK9 in migrating neurons impedes migration, suggesting that proper control of membrane potential/neuronal activity at each developmental step is important for normal cortical development (Bando et al., Cerebral Cortex, 2014; Bando et al., Cerebral Cortex, 2016).
2. How does neuronal activity contribute to cortical circuit construction?
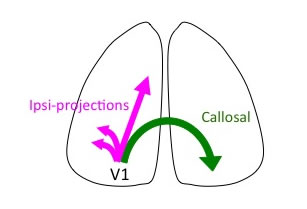
Arriving at their final positions, cortical neurons extend dendrites and axons to form synapses. As synapses are formed between neurons, developing cortical circuits exhibit spontaneously generated, highly correlated neuronal activity. Such synchronous neuronal activity plays important roles in cortical circuit formation. We have found that reducing spontaneous cortical activity by in utero electroporation of an inwardly rectifying K+ channel Kir2.1 impairs callosal axon projections (Mizuno et al., JNS, 2007; Mizuno et al., EJN, 2010; Tagawa and Hirano, Neural Plasticity, 2012). It also disturbs reorganization of orientation selectivity in V1 neurons (Hagihara et al., Nat. Neurosci., 2015). Spontaneous neuronal activity is thus crucial for both long-range axonal projections and local tune-up of circuits in the developing cortex.
3. Mechanism of the regulation of cardiac L-type Ca2+ channel (Cav1.2 channel)
Cav1.2 channel contributes to electrical and contractile function of the heart by controlling the Ca2+ influx through it. The activated intracellular Ca2+ signaling, in turn, controls the activity of Cav1.2 channel.
We are focusing on the mechanisms underlying regulation of Cav1.2 channel by cytoplasmic factors with patch-clamp technique.
[Inside-out patch recording method]
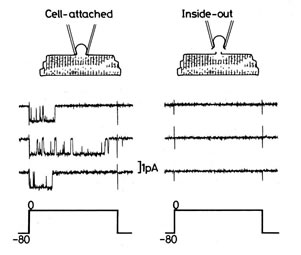
To establish the inside-out recording mode, a pipette is first attached to a membrane patch, as in the cell-attached mode, and then the membrane patch is detached from the rest of the cell. The cytosolic face of the membrane patch is exposed to the external bath solution. Activity of the channel in the patch is analyzed by measuring currents through the channel. One advantage of this method is that we can easily change the composition of solution in the intracellular side of channels or add regulatory factors in the solution according to the research purpose. Thus, this method is particularly valuable for studying influences of intracellular molecules on the function of ion channels.
- ć@ Calmodulin (CaM) and ATP are required for basal channel activity
- Activity of Cav1.2 channel diminishes within several minutes in the inside-out configuration (known as rundown). We have reported that CaM together with ATP reverses rundown of Cav1.2 channel in inside-out patch (Xu et al., 2004), suggesting that CaM and ATP are required for basal channel activity. However, the reversal efficiency of CaM decreases with rundown time. When the channel is in the rundown state for 10 min or longer after patch excision, CaM and ATP fail to restore channel activity. This result suggests that an unknown mechanism blocks the available CaM interaction with Cav1.2 channel. Although CaM is well known to be a ubiquitous and multifunctional Ca2+-binding protein, and mediates the regulation of many cytoplasmic proteins, the mechanism underlying the regulation of Cav1.2 channel by CaM needs to be further clarified.
- ćA Concentration-dependent regulation of Cav1.2 channel by CaM
- Ca2+-dependent facilitation (CDF) and inactivation (CDI) of Cav1.2 channel has been well known. To our surprise, CaM, at a certain [Ca2+], facilitates and inactivates Cav1.2 channel in a concentration-dependent manner (Han et al., 2010), suggesting that CDF and CDI is not only dependent on [Ca2+], but also dependent on CaM concentration.
- Our pull-down assay suggests that a maximal molar ratio of 2:1 for CaM binding to Cav1.2 channel (Asmara et al., 2010; Minobe et al., 2011). Different from the conventional hypothesis on Cav1.2 channel inactivation with 1 molecule of CaM, we have proposed a novel model that 2 molecules of CaM are involved in inactivation of Cav1.2 channel (Han et al., 2010). In this model, a Cav1.2 channel contains 2 CaM binding sites, one site for activation and the other site for inactivation. At a low [Ca2+], 1 molecule of Ca2+-free CaM (apoCaM) binds to the activation site and enhances channel activity. Once [Ca2+] increases to a high level, another Ca2+-bound CaM (Ca2+/CaM) binds to the inactivation site and triggers inactivation of Cav1.2 channel.
- ćB IQ domain is responsible for activation of Cav1.2 channel by CaM
- We have reported that, as well as CaM, L domain of calpastatin (CSL) activates Cav1.2 channel in inside-out patch (Minobe et al., 2006). However, CSL dose-dependently inhibits channel activity induced by CaM, indicating that CSL is a partial agonist of the CaM binding site (Minobe et al., 2011). Furthermore, our pull-down experiments show that CSL competes with CaM for IQ domain, but not for preIQ, suggesting that IQ domain is the activation site of Cav1.2 channel for CaM.
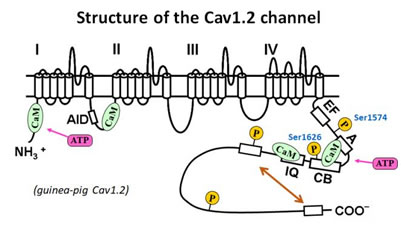
- ćC Phosphorylation of serine 1574 is responsible for protein kinase A (PKA)-mediated facilitation of Cav1.2 channel
- PKA-mediated facilitation of Cav1.2 channel has been well documented, however, the phosphorylation site responsible for the facilitation is controversial. We have examined the effect of forskolin (adenylyl cyclase activator, to elevate cAMP level) on Cav1.2 channels in which different potential PKA phosphorylation sites are mutated. Our result indicates that serine 1574 and 1626 are required for the PKA-mediated facilitation of Cav1.2 channel (Minobe et al., 2014).
- ćD Mechanism of auto-inhibition by distal C-terminus of Cav1.2 channel
- Distal C-terminus (DCT) of Cav1.2 channel has been reported to inhibit Cav1.2 channel via interaction with its proximal C-terminus, known as auto-inhibition. We attempt to clarify the mechanism of auto-inhibition by the experiments with DCT-truncated channels (Minobe et al., 2017) and the fragments of C-terminus (Lyu et al., 2017).
Based on our studies, we think that CaM binding to the channel is crucial for the channel activity. Other cytosolic factors, such as ATP, kinase, Ca2+ and pH regulate the channel activity through modulation of CaM binding directly or indirectly.
- Member
- Professor, Yoshiaki Tagawa
- Lecturer, Jian-Jun Xu
- Lecturer, Etsuko Minobe
- Assistant Professor, Nao Nakagawa
- Link
